Glycogen Storage Disease Type I Glycogen Storage Disease Diet

Glycogen storage diseases Biochemistry notes, Biochemistry, Mnemonics
1. INTRODUCTION. Glycogen storage disease (GSD) type IV (GSD IV, OMIM #232500) is a rare inherited disorder of carbohydrate metabolism first described by Andersen in 1956 as "familial cirrhosis of the liver with storage of abnormal glycogen". 1 The disease is caused by autosomal recessive mutations in the GBE1 gene (OMIM *607839), which leads to 1,4‐α‐glucan‐branching enzyme (ie.

Glycogen Storage Disease Causes, Types, And Treatments Perfect Keto
Glycogen storage diseases (GSDs) are a group of inherited disorders characterized by enzyme defects that affect the glycogen synthesis and degradation cycle, classified according to the enzyme deficiency and the affected tissue. The understanding of GSD has increased in recent decades, and nutrition.
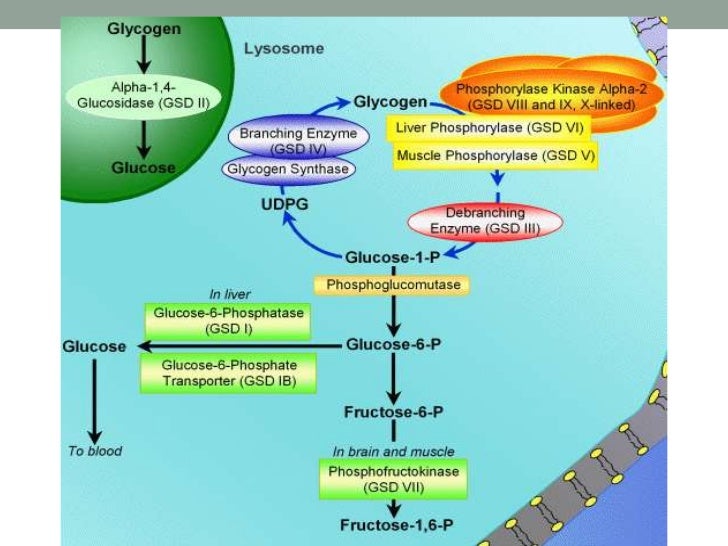
Glycogen Storage Diseases High Yield Notes and Mnemonics
the ingredients. Calcium Supplement and Multivitamin Suggestions in Type I GSD Important Notes about taking Multivitamins and Calcium: Better absorption of the multivitamin occurs when it is taken with food. Do not take more than 500 mg calcium at one time.
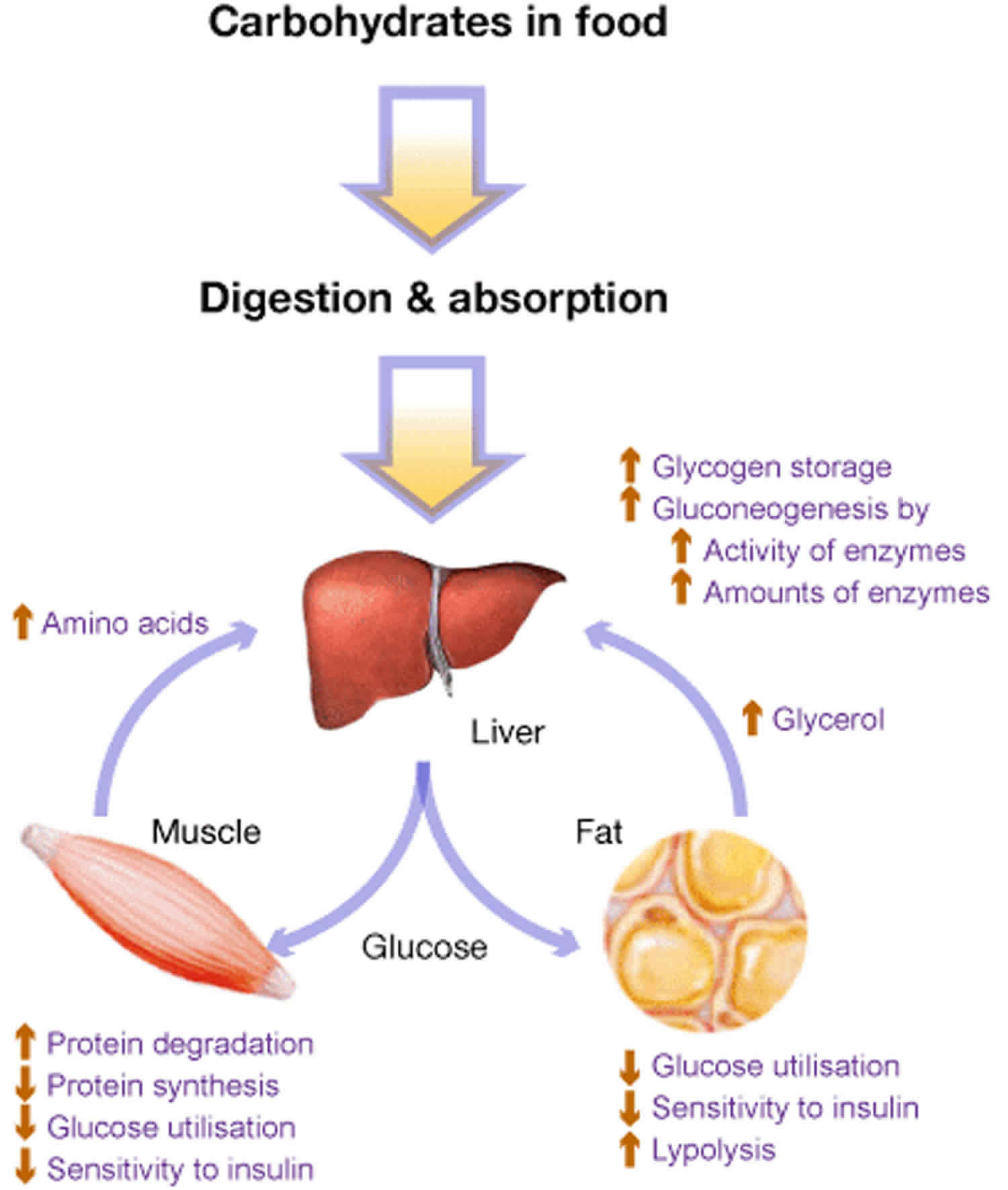
Glycogen Muscle
Disorders of carbohydrate metabolism that result in abnormal storage of glycogen are classified as GSDs. They are classified numerically in the order of recognition and identification of the enzyme defect causing the disorder. Clinical onset can range from neonatal life to adulthood.

GLCOGEN STORAGE DISORDERS
Glycogen storage diseases (GSDs) are a heterogeneous group of inherited disorders caused by inborn errors of glycogen metabolism. These disorders most commonly affect the muscle and liver where glycogen is the most abundant. 1 For GSD I, secondary metabolic disturbances include fasting hyperlactatemia, hyperuricemia, and hyperlipidemia.

Doctors Network Glycogen storage diseases
Glycogen storage disease type Ia (GSDIa; OMIM#232200), also known as von Gierke disease, is an inborn error of carbohydrate metabolism caused by bi-allelic pathogenic variants in the glucose-6-phosphatase gene ( G6PC; OMIM*613742), which accounts for 80% of cases of type I glycogen storage disease [ 1 ].

Glycogen Storage Disease Ppt Bios Pics
Glycogen storage disease type III (GSD III) is characterized by variable liver, cardiac muscle, and skeletal muscle involvement. GSD IIIa is the most common subtype, present in about 85% of affected individuals; it manifests with liver and muscle involvement.. High-protein diet. Protein intake of 3 g/kg or 25% of total energy is recommended.
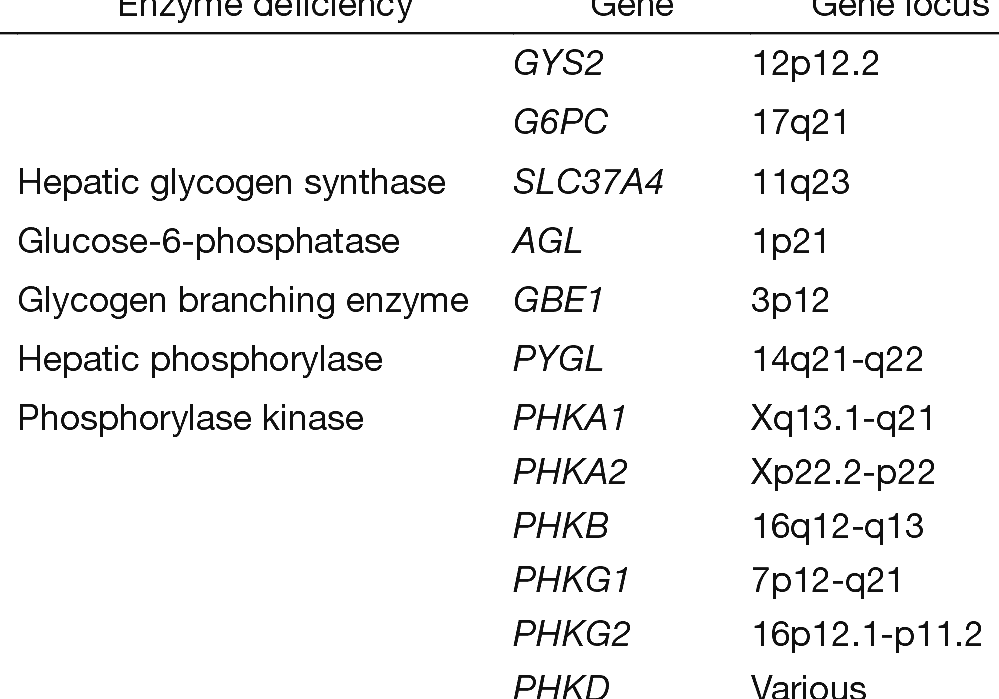
Glycogen Storage Disease Type I Glycogen Storage Disease Diet
The hepatic glycogen storage diseases (GSDs) are a group of disorders where abnormal storage or release of glycogen leads to potentially life-threatening hypoglycemia and metabolic disturbances.. ketogenic diet in GSD I; and 7) supplementation in GSD I. Dietary restrictions. Researchers have known since the 1950s that dietary sucrose.

(PDF) Glycogen storage disease type III modified Atkins diet improves
Glycogen Storage Disease Type IX (GSD IX) is a genetic metabolic disorder which causes the inability to break down glycogen to glucose. Glycogen is a stored form of. You will need to follow a "high protein diet" which is defined as 2-3 grams of protein per kilogram body weight per day. This amount of protein should be spread out

Glycogen storage diseases Download Table
The hepatic glycogen storage diseases (GSDs) are a group of inborn errors of metabolism caused by abnormalities of the enzymes that catalyze the synthesis or degradation of glycogen. The first GSD was described by Edgar von Gierke in 1929 ( 1. ) and there are now at least 16 recognized types ( Table 1 ).

Glycogen Storage Disease Type I StatPearls NCBI Bookshelf
Glycogen storage diseases (GSDs) are a group of rare, monogenic disorders that share a defect in the synthesis or breakdown of glycogen. This Primer describes the multi-organ clinical features of.

Is there any natural treatment for Glycogen Storage Disease?
The hepatic glycogen storage diseases (GSDs) are a group of inborn errors of metabolism caused by abnormalities of the enzymes that catalyze the synthesis or degradation of glycogen. The first GSD was described by Edgar von Gierke in 1929 ( 1) and there are now at least 16 recognized types ( Table 1 ). TABLE 1

Glycogen Storage Disease diet. Is there a diet which improves the
You will need to follow a "high protein diet" which is defined as 3-4 grams of protein per kilogram body weight per day. This amount of protein should be spread out throughout the entire day. For example protein should be consumed at the following times: breakfast, mid-morning snack, lunch, mid-afternoon snack, dinner, and snack before bed.

Glycogen Foods the Best Foods to Increase Your Glycogen Level Human
Glycogen storage disease type III (Cori disease, Forbes disease, amylo-1,6-glucosidase deficiency, glycogen debrancher deficiency) results from deficient glycogen debrancher enzyme activity, which has 2 independent catalytic activities: oligo-1,4-1,4-glucantransferase and amylo-1,6-glucosidase.. In general, an infusion of an elemental diet.
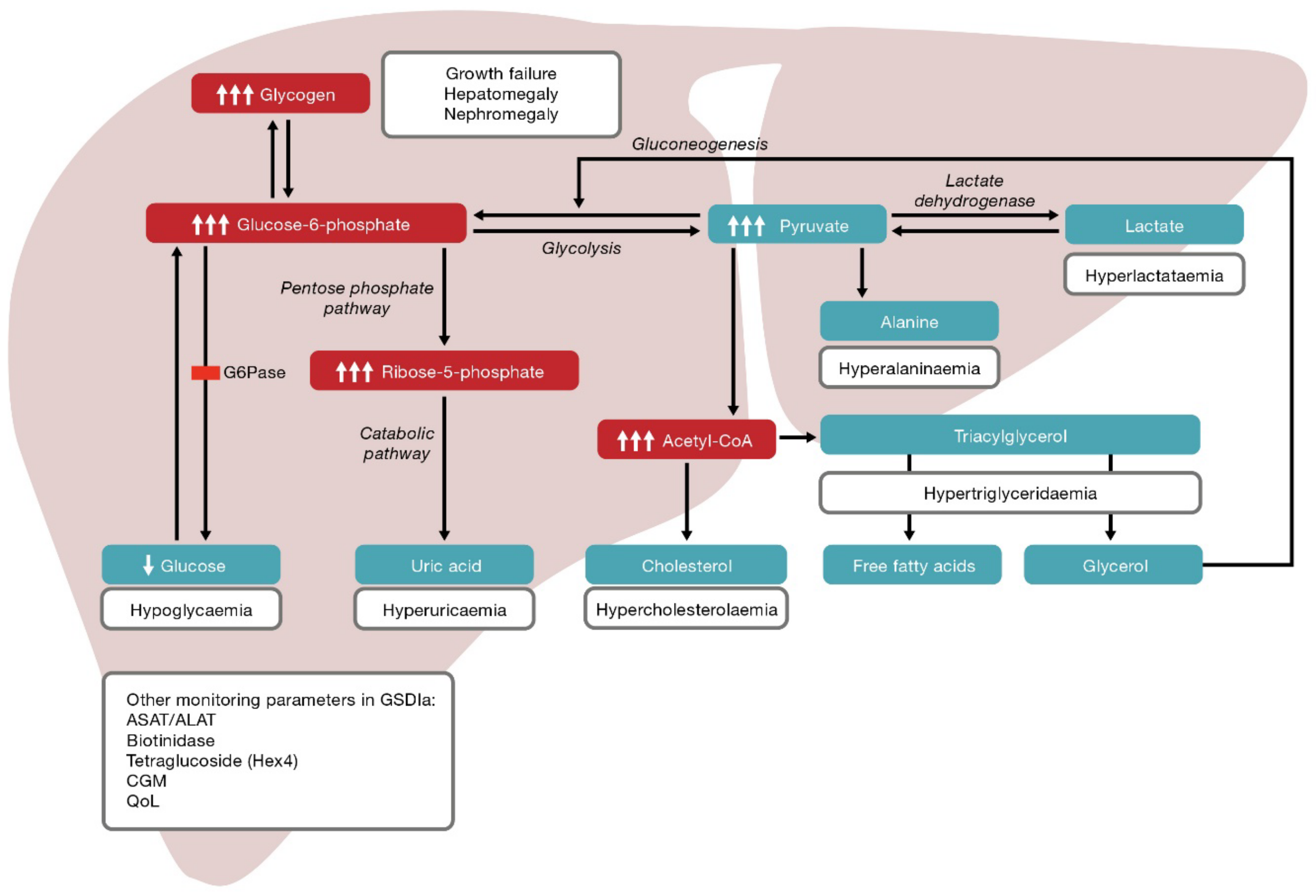
Glycogen Storage Disorders Types Dandk Organizer
Glycogen storage disease (GSD) is a rare metabolic disorder where the body is not able to properly store or break down glycogen, a form of sugar or glucose. GSD affects the liver, muscles and other areas of the body, depending on the specific type.

Glycogen Storage Diseases Nursing School Notes, Medical School, School
Glycogen storage disease Type III (GSD III) is an autosomal recessive disease due to the deficiency of the debranching enzyme, which has two main consequences: a reduced availability of glucose due to the incomplete degradation of glycogen, and the accumulation of abnormal glycogen in liver and cardiac/skeletal muscle.
